Болезнь Хантингтона
Содержание
Определение
Болезнь Хантингтона – аутосомно-доминантное хроническое наследственное нейродегенеративное заболевание.
Классификация
Стадии болезни Хантингтона:
0 стадия – нормальные макроскопические и микроскопические данные;
1 стадия – только гистологические изменения;
2 стадия – атрофические изменения в полосатых телах, по конвексу головок хвостатых ядер;
3 стадия – более выраженная атрофия полосатых тел, уплощение головок хвостатых ядер;
4 стадия – наибольшая атрофия полосатых тел, вогнутость головок хвостатых ядер.

Рис. 1. Схематичное изображение головного мозга в аксиальной плоскости на уровне базальных ядер: указана атрофия хвостатых ядер (узкие стрелки) и расширение конвекса передних рогов боковых желудочков (широкие стрелки).
Патогенез
Предполагаемым механизмом развития заболевания является генетический дефект, который заключается в повреждении короткого плеча 4 хромосомы с формированием белка хантингтина. При развитии заболевания происходит агрегация протеина и его аккумуляция в терминальных отделах аксонов. На аутопсии выявляется генерализованная атрофия мозгового вещества с потерей около 30% его веса. В патологический процесс вовлекается как кора, так и белое вещество гемисфер головного мозга. Наиболее выраженные макроскопическими изменениями являются потеря объёма мозговой ткани с уменьшением головок хвостатых ядер, скорлупы и бледных шаров.

Рис. 2. Изображение аутопсии во фронтальной плоскости, отмечается истончение головок хвостатых ядер (широкие стрелки), расширение конвекса передних рогов боковых желудочков (белые стрелки) и атрофические изменения скорлупы (черные стрелки) (А); истончение головок хвостатых ядер (узкие прямые стрелки) на фоне расширения боковых желудочков (широкие стрелки) и базальных цистерн (изогнутые стрелки) (Б).
Микроскопически болезнь Хангтингтона проявляется потерей нейронов с включениями протеина хантингтина, астроцитарным глиозом и аккумуляцией железа. Изменения более выражены в базальных ядрах, но могут также выявляться в остальных отделах головного мозга включая мозжечок (который менее часто вовлекается во взрослом возрасте).
Эпидемиология
Частота выявления составляет 4-7 случаев на 100.000 в большинстве популяций, менее часто встречается в Азиатских и Африканских популяциях. Средний возраст манифестации заболевания 35-45 лет. Только 5-10% пациентов имеют симптомы заболевания до возраста 20 лет (ювенильная форма заболевания). Ювенильная форма в дополнении к атрофии хвостатых ядер проявляется атрофическими изменениями в мозжечке. В частоте развития заболевания нет гендерной предрасположенности.
Визуализация
КТ, МРТ.
Клинические проявления
Симптоматика зависит от возраста начала появления симптомов. Взрослая форма характеризуется прогрессирующей потерей нормального функционирования моторных нейронов, развитием стереотипичных хореоформных движений и спутанности сознания. С момента возникновения симптомов заболевание неуклонно прогрессирует с летальным исходом в течение 10-20 лет. Ювенильная форма характеризуется ригидностью и дистонией в большей степени чем хореей. Так же выявляются симптомы нарушений со стороны мозжечка.
Лучевая диагностика
КТ-семиотика
Выявляются атрофические изменения в скорлупе, головках хвостатых ядер и в меньшей степени полосатых телах с компенсаторным расширением передних рогов боковых желудочков. Определяется диффузная церебральная атрофия (в некоторых исследованиях указывается на преимущественное вовлечение лобных долей).
Атрофия головок хвостатых ядер измеряется на уровне третьего желудочка:
- увеличение расстояния между медиальными отделами головок хвостатых ядер, при болезни Хантингтона расстояние между головками хвостатых ядер обычно более 20 мм и часто более 25 мм в сравнении с нормой: 10-14 мм.
- расстояние между головками хвостатых ядер можно сравнить с расстоянием между внутренними пластинками черепа, при болезни Хантингтона составляет 0.175-0.185 в сравнении с ~0.12 в норме.
- расстояние между головками хвостатых ядер и латеральными углами передних рогов боковых желудочков, при заболевании варьируется от 1.3 до 1.8 (норма 2.3-2.8).
Контрастного усиления в пораженных структурах не определяется.

Рис. 3. МСКТ в аксиальной плоскости, показана генерализованную атрофию головок хвостатых ядер и полосатых тел с диспропорционально увеличенными передними рогами боковых желудочков (стрелки) (А). МСКТ во фронтальной реконструкции, отмечается увеличение передних рогов боковых желудочков с выпрямлением латеральных границ с увеличением расстояния между головками хвостатых ядер (стрелки) на фоне их атрофических изменений (Б).
МРТ-семиотика
Т1-ВИ: происходит уменьшение головок хвостатых ядер с увеличением расстояния между ними. Уменьшение объёма всех базальных ядер, может выявляться даже до появления первых симптомов. В поздних стадиях наблюдается диффузная церебральная атрофия. Атрофические изменения мозжечка выявляются при ювенильной форме.

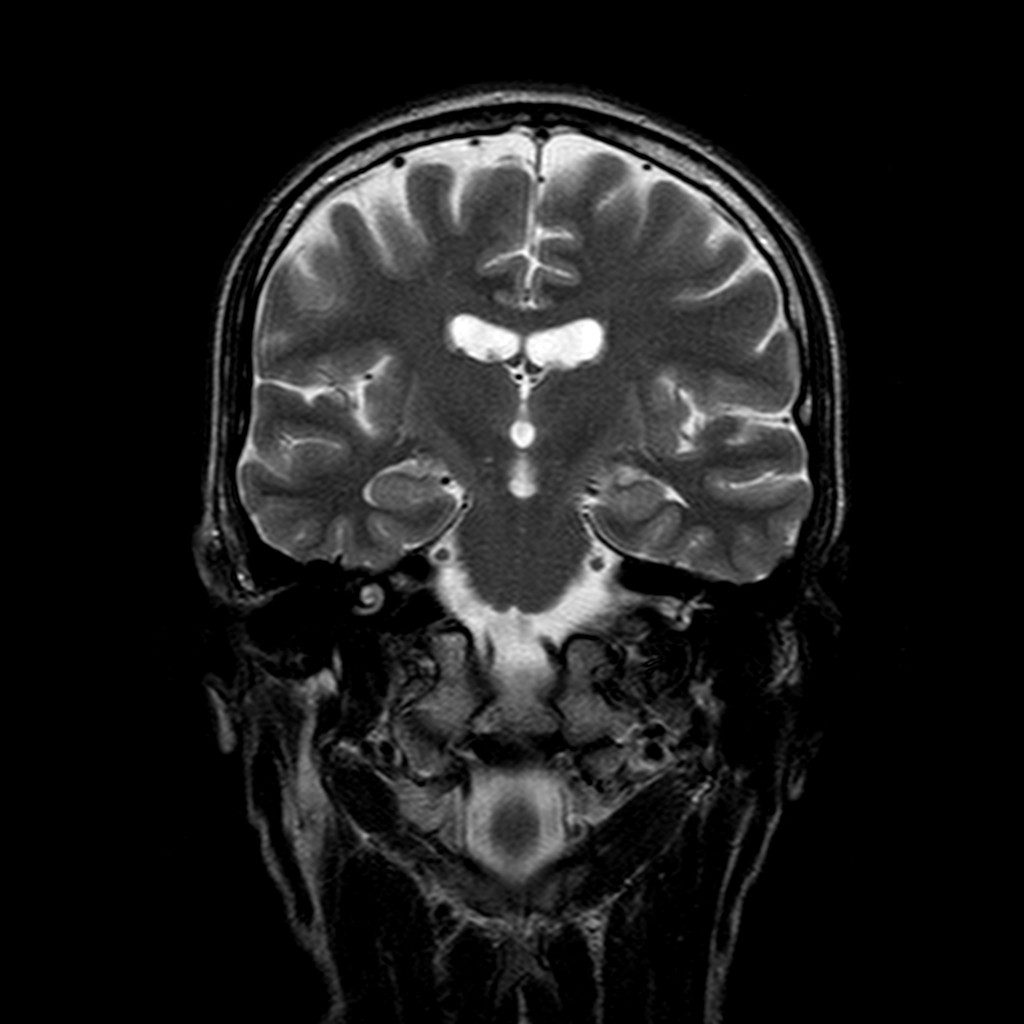
Рис. 4. Т1-ВИ во фронтальной плоскости: показано сравнение расстояния между головками хвостатых ядер в норму (А, Б, В) и пациента с болезнью Хантингтона (Г, Д, Е).
Т2-ВИ: при ювенильной форме может наблюдаться повышение сигнала от головок хвостатых ядер. Возможно определение сниженного сигнала от полосатых тел вследствие отложения депозитов железа.

Рис. 5. Т2-ВИ в аксиальной плоскости: определяется расширение краёв передних рогов боковых желудочков за счет атрофических изменений базальных ядер (прямая стрелка), а также уменьшение размеров полосатых тел с повышением её сигнальных характеристик (изогнутая стрелки) (А); Flair в аксиальной плоскости, отмечается повышение сигнала от головок хвостатых ядер (широкие стрелки) и полосатых тел (узкие стрелки) на фоне их атрофических изменений с компенсаторным расширением передних рогов боковых желудочков (Б).

Рис. 6. Т2-ВИ (А) и Flair (Б) в аксиальной плоскости одного пациента: визуализируются характерные атрофические изменения головок хвостатых ядер (узкие стрелки) и полосатых тел (широкие стрелки) на фоне генерализованных атрофических изменений головного мозга с расширением боковых желудочков и борозд.

Рис. 7. Т2-ВИ (А) и Flair (Б) в аксиальной плоскости у пациента с ювенильной формой болезни Хантингтона: наблюдается повышение сигнала и атрофические изменения полосатых тел симметрично с обеих сторон (стрелки).
DWI: участки пораженных структур не ограничивают диффузию;
T1+C: не определяется патологического накопления контрастного препарата.
МР-спектроскопия: повышение концентрации лактата в коре затылочных долей при наличии симптоматики и в базальных ядрах у некоторых пациентов. Уменьшение соотношения N-ацетиласпартата к креатину в базальных ядрах (как следствие потери нейронов). Выраженное увеличение соотношения холина к креатину в базальных ядрах.
ОФЭКТ: нарушение перфузии в моторной коре, префронтальной коре и базальных ядрах, степень коррелирует клинической картиной.
ПЭТ: уменьшение поглощения препарата в базальных ядрах возникает до отчетливых проявлений атрофии. Гипометаболизм лобных долей.
Дифференциальный диагноз
- Синдром Лэя;
- Болезнь Вильсона;
- Пантотенат-киназа ассоциированная нейродегенерация;
- Отравление углекислым газом;
- Другие нейродегенеративные нарушения.
Пример описания
Описательная часть: Определяется симметричное умеренно выраженное повышение сигнала и умеренные атрофические изменения головок хвостатых ядер и скорлупы в режимах Т2 и Flair с компенсаторным увеличением передних рогов боковых желудочков и расширением расстояния между головками хвостатых ядер до … см, без признаков перифокального отека и ограничения диффузии в режиме DWI. На постконтрастных изображениях признаков
ЗАКЛЮЧЕНИЕ: МР-картина структурных, умеренных атрофических изменений головок хвостатых ядер и скорлупы, с компенсаторным расширением передних рогов боковых желудочков (МР-семиотика специфична для проявлений болезни Хантингтона).
Список использованной литературы и источников
- Brain imaging with MRI and CT: an image pattern approach / [edited by] Zoran Rumboldt et al. 1st edition. Cambridge University Press, New York, 2012. – P. 415. ISBN: 978-0-521-11944-3.
- Diagnostic imaging. Brain / [edited by] A. Osborn, Karen L. Salzman, and Miral D. Jhaveri. 3rd edition. Elsevier. Philadelphia, 2016. – P.1213. ISBN: 978-0-323-37754-6.
- Magnetic resonance imaging of brain and spine / [edited by] Skott W. Atlas. 5th edition. Walters Kluwer. Philadelphia, 2017. – P.2257. ISBN: 978-1-469-87320-6.
- MR Neuroimaging: Brain, Spine, Peripheral Nerves / [edited by] Michael Forsting, Olav Jansen. 2nd edition. Georg Thieme Verlag KG, Stuttgart, 2017. – P.582. ISBN: 978-3-13-202681-0.
- Osborn’s brain / [edited by] A. Osborn. 2nd edition. Elsevier. Philadelphia, 2017. – P.1372. ISBN: 978-0-323-47776-5.
|